 当前位置:
当前位置:随着新一代测序技术的发展和全基因组测序(Whole Genome Sequencing, WGS)成本的不断下降,基于WGS的分离群体分组分析(Bulk Segregation Analysis,BSA)已经成为快速定位候选基因的常规工具,但已报道的方法依然有待完善。例如,依赖于自交系杂交的QTL-seq策略,需要后期耗费大量精力进行精细定位;而基于突变体的Mutmap策略,虽然不需要后期进行精细定位,但依然需要耗费大量时间用于突变体的自交与回交。这些方法的不足限制了大规模开展功能基因定位的效率。
中国科学院东北地理与农业生态研究所大豆分子设计育种重点实验室冯献忠团队,围绕大豆突变体功能基因定位,报道了一种称为M2-seq的改良版WGS-BSA方法。它是一种快速有效的突变基因定位工具,仅基于M2代材料即可实现候选因果突变位点的快速定位,与以前报道的需要更高世代自交与回交的方法(例如,Mutmap)相比,在时间成本和测序费用成本上都更具有优势。利用M2-seq方法,在不知道突变植株的野生型变异信息情况下,通过M2群体之间的相互比较,背景变异即可被有效地去除。此外,利用 ΔSNP-index的绝对值可以有效去除由相邻突变等位基因的排斥连锁引起的突变频率信号波动,从而有助于定位靶基因中的因果突变。本研究展示了M2-seq在10个独立的大豆M2突变体群体中成功定位因果突变。研究表明,基于M2世代的M2-seq方法可以加速基因定位,尤其是在世代间隔较长的植物物种中。
本研究以“A robust and rapid candidate gene mapping pipeline based on M2 populations”为题在国际期刊 Frontiers in Plant Science发表。东北地理所博士研究生周煌凯为论文第一作者,冯献忠研究员为论文通讯作者。相关研究得到了国家重点研发计划资助。
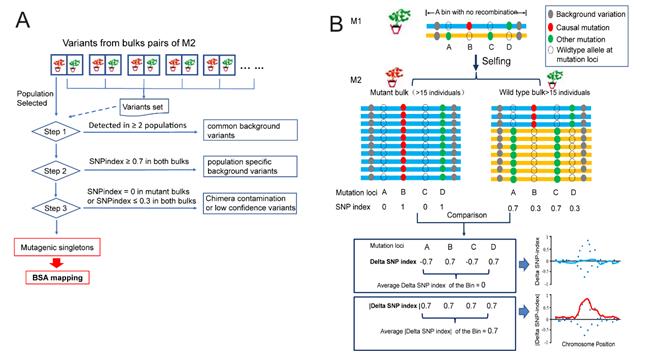
图1 M2-seq的原理和分析过程示意图
(A) 变异过滤过程概述。(B) 定位携带因果突变的基因组区域的原理图。ΔSNP-index绝对值曲线(红色曲线)用于识别因果突变区域。

 |
 |